| Reading a structure optimization output file
MOLDRAW reads the .OUT output CRYSTAL03 file resulting from single point energy calculation
and structure optimization (unit cell parameters and/or atomic fractional
coordinates) calculations by the Imp
CRY03 keyword of the MOLDRAW toolbar.
Once a CRYSTAL03 structure optimization file is read, each structure is saved
as a CRY_nn.MOL file in the TMP_P directory of the local directory. The
nn digit identifies the nn structure in
the optimization process.
MOLDRAW steps through each structure to follow the change in geometry
and unit cell parameters due to energy minimization. The reported
total energy always refers to the unit cell content.
For the crystalline systems, either bulk or slab, are supported. A
geometry optimization sequence can be re-opened without converting again the
CRYSTAL03 output file by the (File--->Open sequence...) menu
and selecting the file CRY_SEQ.SEQ which is located in the TMP_P
directory.
|
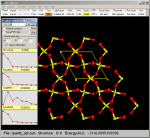
|
|
The forward/backward buttons allow to view the sequence of the
structures in a continuos process, as in a movie.
Each structure is also saved as a MOL file in the TMP_P
subdirectory. For instance, if the name of the original CRYSTAL file
is quartz.out and three structures
are present as a result of the optimization then three files named
cry_01.mol, cry_02.mol and cry_03.mol are written in the TMP_P
directory. |
|
|
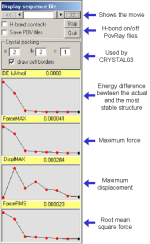
|
Four ASCII files are also written called:
Quartz.out_DISPLMAX_graph.dat
Quartz.out_ENERGY_graph.dat
Quartz.out_FORCEMAX_graph.dat
Quartz.out_FORCERMS_graph.dat
each containing the corresponding data from the ab-initio calculation
in a format ready to be used by any plotting programs.
A given structure can also be visualized by clicking each red dot on
any curve.
Hydrogen bond contacts can be turned on/off by selecting the proper
option button.
In this case a larger portion of the quartz crystal has been chosen
(a=3, b=2, c=1) and cell border have been included.
|
The various structures in the sequence can also be saved as PovRay files to be processed by the PovRay program. In this case
files named cry_01.pov, cry_02.pov to cry_09.pov are written in the TMP_P
directory to be processed by the PovRay program. To see how to trasform a
sequence of PovRay files in an AVI movie please click this link.
A geometry optimization sequence can be re-opened without converting again
the CRYSTAL output file by the (File--->Open
sequence...) menu and selecting the file CRY_SEQ.SEQ file
located in the TMP_P directory.
You are here: Home-Navigate-Topics-CRYSTAL-Optimization
Previous Topic: Export input Next Topic: Frequencies
| 

